![]()

MEDICAL TEAM MAGAZINE
Anno 9 - Numero 1 - gen/apr 2010

Daniela Concolino
Ricercatore della Cattedra di Pediatria
Università Magna Graecia di Catanzaro.
Referente del Centro Regionale
Malattie Rare Pediatriche Regione Calabria
Anno 9 - Numero 1
gen/apr 2010
| LA SINDROME
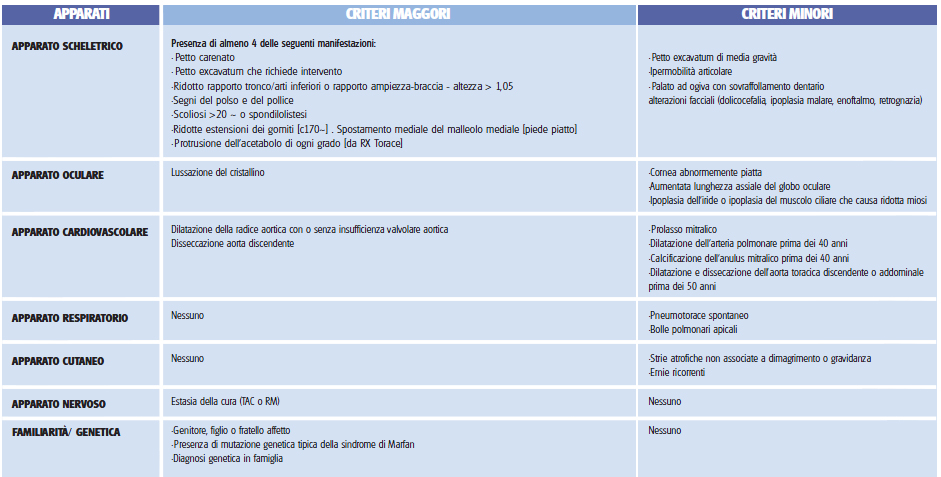
DI MARFAN LA SINDROME DI MARFAN è una malattia del tessuto connettivo a trasmissione autosomica dominante la cui prevalenza è stimata intorno a 1/5000 persone. Il paziente affetto da sindrome di Marfan è classicamente un soggetto molto alto e magro, con una conformazione particolare delle dita della mano che risultano affusolate e molto lunghe [aracnodattilia], che spesso manifesta una iperlassità articolare, ovvero la capacità di compiere movimenti innaturali delle articolazioni come iperflettere le dita, i gomiti o le ginocchia, e delle alterazioni della gabbia toracica [petto escavato o carenato] o della colonna vertebrale [scoliosi]. Non di rado però la presentazione clinica del soggetto affetto si discosta dalla manifestazione classica della patologia e non è raro trovare soggetti che non presentano tutto il corteo sintomatologico sopra descritto. Per valutare il sospetto diagnostico di Sindrome di Marfan attualmente è utilizzato uno score definito Ghent nosology [tab1]. Questo si basa sulla ricerca di alcuni parametri differenziati in criteri maggiori e minori. Lutilizzo di questi criteri determina o esclude la diagnosi di Marfan nel 86% dei casi. Come si può notare dalla tabella la patologia si evidenzia principalmente in tre distretti che sono locchio, lo scheletro e il sistema circolatorio ma la prognosi è determinata dalla gravità delle complicanze cardiache e circolatorie. Particolare attenzione bisogna porre alla possibilità di un aneurisma o di una dissecazione aortica, che va valutata con periodiche visite ecocardiografiche, e qualora presenti, tempestivamente trattate con lutilizzo di farmaci antipertensivi [beta-bloccanti] o con terapia chirurgica. La diagnosi e il follow-up della malattia si avvalgono di una gestione multidisciplinare in cui è importante che collaborino il medico genetista, il cardiologo, loculista, lortopedico e il reumatologo. CRITERI CLINICI PER LA DIAGNOSI DI SINDROME DI MARFAN [GHENT NOSOLOGY] NOTIZIE UTILI GENETICA
|